Leaves & leaf sample preparation
- 3 Minutes to read
- Print
- DarkLight
- PDF
Leaves & leaf sample preparation
- 3 Minutes to read
- Print
- DarkLight
- PDF
Article Summary
Share feedback
Thanks for sharing your feedback!
Overview
You can send leaves and leaf tissue to us for analysis. While leaves are the most common sample for plants, how you need to send your leaf samples to us depends on whether you are sending your samples from within or outside of Australia. You will also need to ensure that your:
- Samples are consistently sized and within the recommended sizes of 3-5mm for discs or 5-10mm for manually cut samples, and;
- You send enough sample tissue based on the mg weight of the sample.
Sending leaves from within Australia
We can accept 20-25mg of fresh leaves, or 10-15mg if they are lyophilised (freeze dried in a vacuum) or oven dried. If you are sending samples from within Australia, you do not need to pulverise your samples.
Sending leaves from outside Australia
We can only accept 10-15mg of tissue that has been lyophilised (freeze dried in a vacuum) or oven dried.
Please note, if sending samples from outside Australia, your samples must also be pulverised to ensure they pass Australia’s strict quarantine laws. See the Australian Border Force website for more information.
We recommend you collect leaves into liquid nitrogen, but if not, collect them onto ice blocks or, if that’s also not possible, a desiccant like silica beads or gel.
Important
Please do not send leaf material at room temperature that has previously been frozen. The tissue may decompose and the DNA will be degraded before it can reach us.
How to prepare leaves and leaf samples
Step 1: Cutting leaves and leaf samples
To prepare your leaves or leaf samples for analysis you need to provide 20-25mg (if sending from within Australia) of fresh plant tissue per tube. Depending on your sample type, there are two recommended methods for cutting leaves.
Method A: Tube cutting leaf samples
This method is suitable for most leaves excluding acicular or linear leaves. With this method, you can easily cut leaves into equal size 3-5mm discs by:
- Folding the leaf twice

- Using the open-end of the collection tube as a stencil to cut the discs
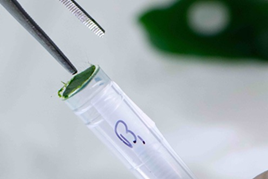
- Pushing the disc down into the tube. You should put 2-3 leaf discs of 3-5mm diameter in each tube tube. Leave tubes G12 and H12 empty as these are used for control purposes.
- Fresh leaf samples must be shipped by express post or overnight courier company (e.g. DHL or Fedex) to prevent moisture build-up inside the tubes that will enhance microorganism growth. If shipping by standard post, oven dry or use silica beads/gel to extract the moisture from the tissue.



Method B: Manually cutting leaf samples
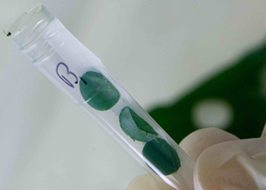
You can also manually cut leaves into 5-10mm pieces, but you will need to ensure that the sample sizes are uniform and consistent across all tubes. Cut the leaves with scissors or a blade, wiped clean with 70% ethanol between samples, to avoid cross contamination. This method ensure the leaves will be pulverised efficiently for successful DNA extraction.
Important
Ensure you maintain consistent sample sizes across all tubes.
The image below shows an example of accurately manually cut leaf samples.

Step 2: Labelling tubes
- Label the tubes with their well position within the plate
- Plates are arranged in columns 1 to 12, and rows A to H
- Firmly and securely seal all the tubes across the rack, making sure the labelled tubes are positioned in the correct plate orientation

Important
Avoid mixing different tissue types (e.g. leaves and roots) within a single plate as each may require a different processing method and do not mix species of a different genus in the same plate. You must create separate orders for each genus.
Unacceptable sample sizes
Please ensure each tube has equal and consistent size samples per our guidelines. In the example below, samples B1 to H1 are not able to be effectively crushed for DNA extraction because they are inconsistently sized and have not been cut correctly. If we need to sub-sample manually, this may affect the cost and time it takes to process your order.

Was this article helpful?
